This cloud computing research success story concerns a large computation. This is generally known as High Performance Computing (HPC) or
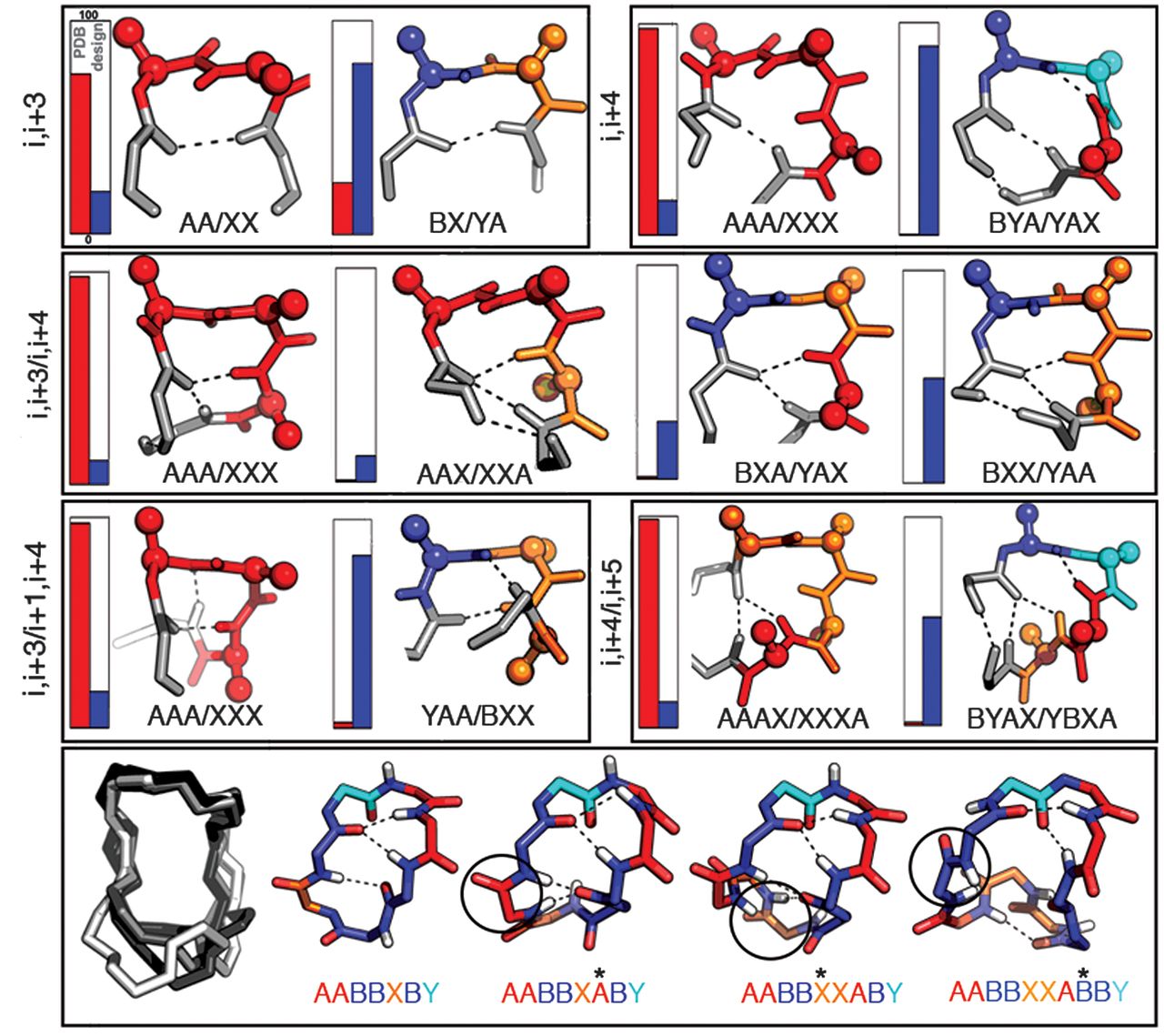
The research objective was to explore a combinatoric space of small proteins (peptides) built from chains of ten mixed-chirality amino acids. There are 39 of these so the search space is 3910 possible peptides. The technical objective was to distribute the computing task across many virtual machines (VMs) on the Amazon Web Services (AWS) public cloud. The computations used the Rosetta 'protein folding' molecular design software to resolve the likely structure of a given amino acid chain. When Rosetta finds that a candidate protein folds into a stable closed loop it is called a macrocycle scaffold. Such stable proteins are interesting and potentially desirable because of their possible applications in therapeutic medicine.
 |
| Figure "Recurrent local structural motifs in designed macrocycles" from Comprehensive computational design of ordered peptide macrocycles |
This case study shows that a moderately large
This documented success, replicating VMs on the AWS public cloud can be applied to research computing tasks that rely on many independent computational threads. Technology components of this study include:
- The Baker Lab Rosetta software suite
- Powerful EC2 compute instances
- The AWS Spot market
- Optimization analysis of cloud computing instance types
- The AWS Batch service
- The AWS Research Credit Grant program
Computing at Scale on AWS
The following steps can help generalize this work and apply it to other parallel computation tasks:
- Identify the research problem that requires large-scale computing
- Configure an AWS User account
- Configure the execution software and the data structure
- Configure a cluster management service such as AWS Batch to run the job at scale on the AWS Spot Market
- The Spot Market provides VMs at typically one-third of the normal cost with a low but non-zero probability that they will be interrupted during execution. Using these instances reliably extends the budget by a factor of three.
- Recover the results and dismiss the
compute infrastructure
The computation in more detail:
- 164 C4.8XLarge instances ran for 53 hours on a single
compute task producing 5.2 million positive-result protein structures after 313,000 virtual CPU hours, documented on GitHub here; - The $0.40 Spot Market cost per instance-hour showed no significant cost variation over the task duration, nor impact on market price;
- Optimization by the researcher saved more than $600 over other instance choices, and the total computation cost came to $3,477.
- Related links:
Cost tradeoffs
The traditional approach to HPC includes purchasing and maintaining dedicated hardware, such as the
 |
Hard Break-even
Soft Break-even
In this case study, the research team had access to an on-premise cluster that was shared by many researchers. This resource would ideally complete the processing task in two weeks, although the shared nature of the cluster means both uncertainty and hassle, so the researcher chose to use AWS. This
Science background
The research in this case study contributed new information on how to tackle the challenge of large-scale sampling of peptide scaffolds, which could help with developing new therapeutics.
Human DNA consists of 3 billion base molecules that are arranged in pairs as rungs of a helical ladder. The base pair sequence records how to construct proteins: each base (nucleotide) can have one of four abbreviated values: A, C, G or T. Three bases in a row can be thought of as three digits in base-4; for example AAG or TCA. That is, this triple is a number from 0 to 63. These triples map to one of 20 left-handed amino acids (with some values degenerate and others not assigned.)
Amino acids are 20 naturally-occurring molecules that are the building blocks of proteins and all life on Earth. 19 of these amino acids have a “left-handed” asymmetry and can be artificially manufactured in their mirror image, producing a total of 39 amino acid building blocks. Peptides are proteins made of small chains of these acids. Once a particular sequence of amino acids is bonded together end-to-end, its rotational degrees of freedom permit it to fold into a structure with favorable energy; this structure may serve some chemical or biological function. The Rosetta software can analyze the manner of this folding, thereby connecting a hypothetical amino acid sequence to a protein scaffold.
One of the practical challenges is matching these structures to naturally-occurring geometries within the organism’s molecular landscape. This is often described using a ‘lock and key’ analogy: a therapeutic molecule could be designed to fit a particular binding site, like a cell wall, which would partially enhance or restrict the metabolic process at that location. The goal would be to comprehensively sample all possible shapes, creating a number of highly stable ‘keys’ that are ready to be slightly re-configured into any desired shape.
The
The research team would like to express their gratitude to Amazon Web Services for assistance in this exploratory project in the form of cloud credits to defray expenses.
